
The degeneracy of a primer is the number of unique sequences it corresponds to (6 in the example above). Degenerage primers can be used in PCR reactions to amplify many related sequences from genomic DNA or from cDNA libraries. They can be used when some of the related genomic sequences are unknown, or known only in a related species.
In expriments, pairs of primers with combined degeneracies of up to 1010 were successfully used to amplify sequences from genomic background, with specificity over 99.5% [1].
In the example below, a pair of primers of length 6 cover a set of 5 sequences.
Notice that there is a mismatch between the 3' primer and the fourth sequence.
| Phase 1: | Locate conserved regions in the DNA sequences by finding ungapped local alignments with a low entropy score. |
| Phase 2: | Design primers using variants of simple approximation algorithms, called CONTRACTION and EXPANSION (these algorithms approximate the number of sequences the primer does not match, provided that the sequences are over a binary alphabet). |
| Phase 3: | Run a greedy hill-climbing procedure to improve the primers, and select the one with the largest coverage as the output. |
The full details of the algorithm and the relevant computational analysis
are described in [2,3].
HYDEN is written in C++, and runs under Windows and Linux.
The file output.txt contains a sample output of HYDEN for this input file. The output shows two pairs of primers of length 25 and degeneracy ~5,000 (5' end), ~30,000 (3'). The primers cover 40 out of the 50 input genes, with up to 3 mismatches (in both ends combined) per covered gene.
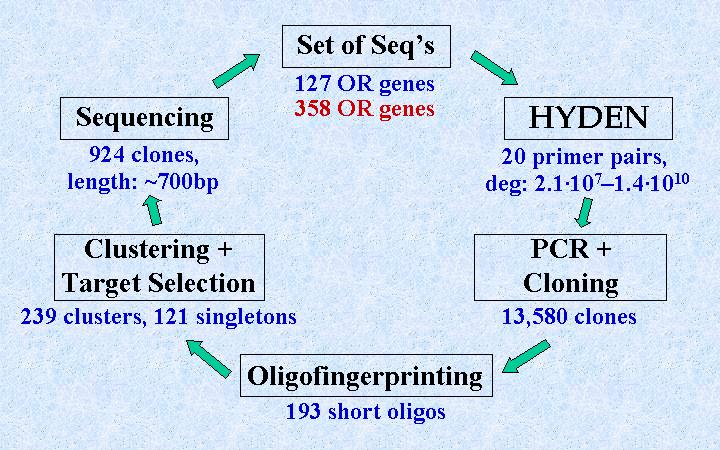
Given a subset of known gene sequences, HYDEN is used to design degenerate primer pairs. The primers are then used in PCR procedures to amplify fragments of genes, known as well as unknown, of the same family. The fragments are cloned, and an oligofingerprinting (OFP) process [4] characterizes the clones by their patterns of hybridization with a series of very short (8-mer) oligonucleotides. The hybridization pattern of a clone is called its fingerprint. Another novel algorithm, called CLICK [5], clusters the clones into groups corresponding to the same gene according to their fingerprints. Finally, representatives from each cluster are sequenced and compared to the existing OR database. The newly revealed sequences can be used to design primer pairs for another cycle of DEFOG.
The DEFOG scheme is illustrated below. The numbers beneath the boxes summarize the actual parameters in our DEFOG experiment on the human OR subgenome.
| [1] | T. Fuchs, B. Malecova, C. Linhart, R. Sharan, M. Khen, R. Herwig,
D. Shmulevich, R. Elkon, M. Steinfath, J.K. O'Brien, U. Radelof, H. Lehrach,
D. Lancet, and R. Shamir, "DEFOG: A Practical Scheme for Deciphering Families
of Genes",
Genomics, Vol. 80, No. 3, pp. 295-302, 2002. pdf. |
| [2] | C. Linhart and R. Shamir, "The Degenerate Primer Design Problem",
Bioinformatics, Vol. 18, Suppl. 1, pp. S172-S180, 2002: pdf. |
| [3] | C. Linhart and R. Shamir, "The degenerate primer design problem: Theory and applications",
JCB, Vol. 12(4), pp. 431-456, 2005: pdf. |
| [4] | U. Radelof, S. Hennig, P. Seranski, M. Steinfath, J. Ramser, R.
Reinhardt, A. Poustka, F. Francis, and H. Lehrach, "Preselection of Shotgun
Clones by Oligonucleotide Fingerprinting: An Efficient and High Throughput
Strategy to Reduce Redundancy in Large-Scale Sequencing Projects",
Nucleic Acids Research, Vol. 26, pp. 5358-5364, 1998. |
| [5] | R. Sharan and R. Shamir, "CLICK: A Clustering Algorithm with Applications
to Gene Expression Analysis",
Proc. 8th International Conference on Intelligent Systems for Molecular Biology (ISMB 2000), pp. 307-316, 2000: ps. |